-
YTHDF2通過識別m?A修飾的U6 snRNA調控炎癥反應和腫瘤發(fā)生的新機制
發(fā)布時間: 2025-11-21 點擊次數(shù): 6次
研究背景:
RNA的N6-甲基腺苷(m?A)修飾是真核生物中普遍的內部RNA化學修飾,被譽為“表觀轉錄組學"的核心調控層。過去十年的大量研究揭示了m?A在信使RNA(mRNA)的代謝、剪接、翻譯和降解中扮演關鍵角色,并通過其“閱讀蛋白"(如YTHDF家族)執(zhí)行功能,進而廣泛影響細胞生理與病理過程,包括腫瘤發(fā)生。YTHDF2是其中重要的閱讀蛋白,其主要功能是識別m?A修飾并促進靶標mRNA的降解。
然而,絕大多數(shù)研究集中于mRNA和長鏈非編碼RNA,對于m?A修飾在種類繁多的“自我"非編碼RNA(尤其是具有核心細胞功能的snRNA)上的作用知之甚少。U6 snRNA是剪接體的關鍵組成部分,傳統(tǒng)認知中其功能嚴格限定在細胞核內的pre-mRNA剪接。近年來,研究發(fā)現(xiàn)U6 snRNA在癌細胞中高表達,且可被METTL16進行m?A修飾,但其在細胞質中的命運和生理病理功能是一個未解之謎。
同時,慢性炎癥是多種癌癥的關鍵驅動因素,而紫外線(UVB)是誘導皮膚炎癥和皮膚癌的主要環(huán)境因素。盡管已知Toll樣受體3(TLR3)能識別外源雙鏈RNA引發(fā)免疫反應,但其如何被內源性RNA激活并參與腫瘤發(fā)生,機制尚不明確。本研究正是在此背景下,探索m?A修飾的自我非編碼RNA(以U6 snRNA為代表)是否以及如何通過YTHDF2閱讀蛋白,調控天然免疫應答和腫瘤發(fā)生,從而連接表觀轉錄組學、炎癥與腫瘤三大領域。
研究方法:
本研究采用多層次實驗方法以闡明YTHDF2通過m?A U6 snRNA調控炎癥與腫瘤的機制。
在分子與細胞水平,研究者利用RNA干擾、基因敲除與過表達技術,結合RNA-seq和qPCR,旨在驗證YTHDF2缺失對炎癥通路的關鍵作用。通過m?A-seq、RNA免疫沉淀和體外結合實驗,旨在發(fā)現(xiàn)并證實YTHDF2直接結合m?A修飾的U6 snRNA并介導其降解。使用TLR3抑制劑、基因敲低及LRR結構域突變體,旨在證明U6通過TLR3的LRR21域激活炎癥。
在亞細胞定位層面,采用內體分離、免疫熒光和FISH技術,旨在揭示YTHDF2與U6通過SIDT2轉運至內體并發(fā)生功能互作。
在動物模型與臨床關聯(lián)層面,構建皮膚特異性YTHDF2敲除小鼠并進行UVB致癌實驗,旨在證實YTHDF2在活體水平抑制炎癥與腫瘤發(fā)生。通過分析人類皮膚癌組織芯片與自身免疫病數(shù)據(jù)庫,旨在闡明YTHDF2表達的臨床相關性。
多種方法層層遞進,揭示從RNA修飾到免疫激活的全新信號軸。
主要研究結果:
1、YTHDF2控制炎癥基因表達
研究通過RNA-seq和體內外實驗證實YTHDF2是炎癥的關鍵負調控因子。在人類角質形成細胞中敲低YTHDF2會顯著上調TNF、IL-17和TLR3等炎癥通路相關基因。在構建的皮膚特異性YTHDF2敲除小鼠模型中,UVB照射誘導了更嚴重的皮膚炎癥,表現(xiàn)為表皮增厚、CD45+免疫細胞浸潤增加。在細胞水平,敲低YTHDF2增強了UVB誘導的TNF-α、IL-6、COX-2等關鍵炎癥因子的表達,而過表達YTHDF2則抑制這一過程。此外,臨床數(shù)據(jù)分析發(fā)現(xiàn),YTHDF2在系統(tǒng)性紅斑狼瘡和I型糖尿病患者中表達下調。這些結果共同確立了YTHDF2在抑制炎癥反應中的核心地位。
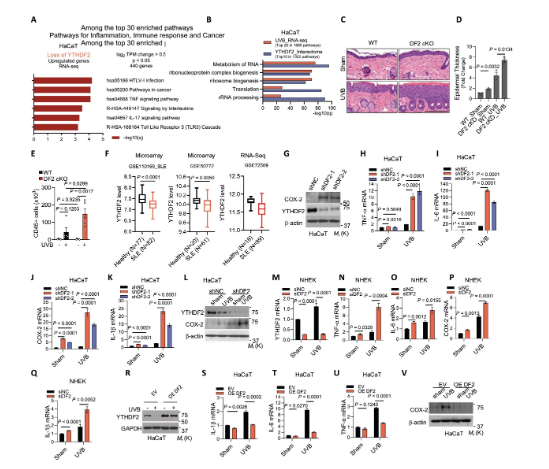
圖1 YTHDF2控制炎癥基因表達
2、YTHDF2結合m?A修飾的U6 snRNA并誘導其降解
為探究YTHDF2調控炎癥的機制,研究發(fā)現(xiàn)了其非編碼RNA新靶點——U6 snRNA。雖然m?A-seq未找到典型的炎癥基因靶標,但通路分析提示非編碼RNA代謝重要。質譜分析顯示YTHDF2與多個U6 snRNP蛋白互作。后續(xù)實驗證實,YTHDF2敲低或UVB照射均能顯著提升U6 snRNA水平,而過表達YTHDF2則降低其水平。通過RIP和m?A-IP實驗,證明YTHDF2直接結合U6 snRNA,且該結合依賴于METTL16催化的m?A修飾。RNA穩(wěn)定性實驗表明,YTHDF2通過促進U6降解來調控其豐度,揭示了其超越mRNA代謝的新功能。
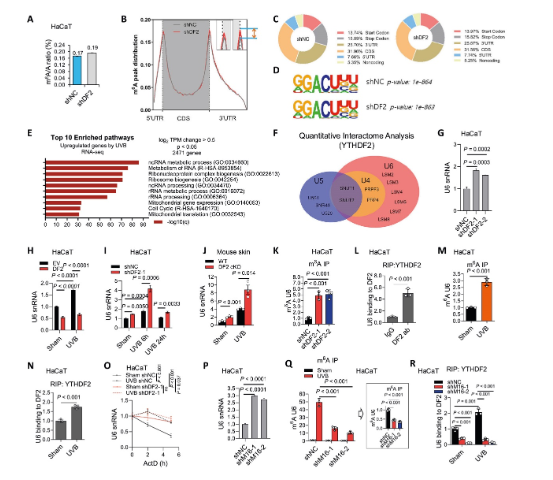
圖2 YTHDF2結合m?A修飾的U6 snRNA并誘導其降解
3、YTHDF2通過U6 m?A甲基化控制炎癥基因表達
研究通過功能挽救實驗證實,YTHDF2調控炎癥依賴于U6 snRNA。在HaCaT細胞中,敲低U6能夠逆轉因YTHDF2敲低引起的TNF-α、IL-6等炎癥因子上調。值得注意的是,體外合成的U6 RNA經(jīng)UVB照射后并不能誘導炎癥,說明其效應并非由UVB直接損傷引起,而是依賴于其內在序列或修飾。METTL16敲低在A431細胞中模擬了YTHDF2敲低的促炎效應,而此效應同樣可被U6敲低所挽救。在YTHDF2敲除的細胞中,METTL16敲低不再進一步加劇炎癥,證明YTHDF2位于METTL16的下游執(zhí)行功能。這些數(shù)據(jù)構建了METTL16-m?A U6-YTHDF2軸調控炎癥的清晰路徑。

圖3 YTHDF2通過U6 m?A甲基化控制炎癥基因表達
4、YTHDF2與m?A U6結合從而抑制其與TLR3的結合
研究闡明了YTHDF2抑制炎癥的精確分子機制:與TLR3競爭性結合U6。小規(guī)模siRNA篩選發(fā)現(xiàn)TLR3是介導YTHDF2敲低促炎效應的關鍵受體。體外結合實驗表明,重組TLR3可同等結合U6與m?A U6,而YTHDF2優(yōu)先結合m?A U6。在細胞內,YTHDF2的存在使m?A U6與之結合,而非TLR3;一旦YTHDF2缺失,m?A U6則轉向與TLR3強力結合,激活炎癥。進一步通過TLR3截斷體鑒定出U6結合其LRR21結構域,而已知的dsRNA類似物poly(I:C)結合的是LRR20域。這揭示了TLR3通過不同結構域區(qū)分不同RNA配體,而YTHDF2通過“占據(jù)"m?A U6來阻斷其激活TLR3。

圖4 YTHDF2與m?A U6結合從而抑制其與TLR3的結合
5、U6與YTHDF2均定位于內體
研究揭示了U6-TLR3通路激活的細胞器定位。細胞組分分離和免疫熒光實驗證實,UVB照射或YTHDF2敲低會顯著增加U6在細胞質和內體中的聚集,并與內體標志物Rab7共定位。YTHDF2同樣被證實存在于內體中。機制上,抑制內吞作用的關鍵蛋白動力或敲低RNA轉運蛋白SIDT2,均能減少U6和YTHDF2在內體中的富集,并降低U6穩(wěn)定性。重要的是,U6敲低會減少內體中的YTHDF2,而YTHDF2敲低則增加內體U6,說明YTHDF2是跟隨U6進入內體的。使用動力抑制劑Dynasore可抑制炎癥基因表達,證明U6進入內體是其激活TLR3所必需。
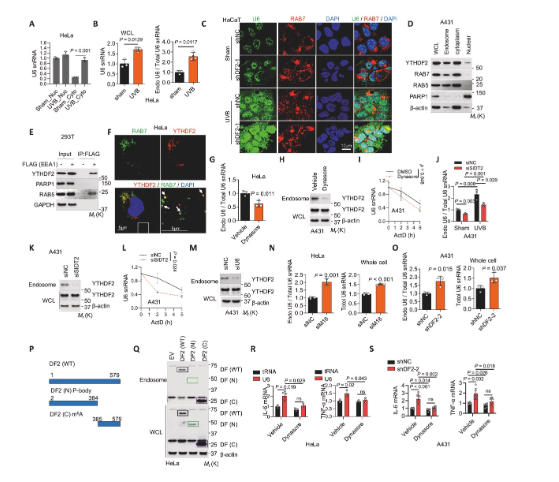
圖5 U6與YTHDF2均定位于內體介導炎癥反應
6、UVB irradiation抑制YTHDF2
研究揭示了UVB通過翻譯后修飾和蛋白降解兩條路徑抑制YTHDF2功能。質譜分析發(fā)現(xiàn)UVB照射后YTHDF2第39位絲an酸發(fā)生去磷酸化。Co-IP實驗表明,去磷酸化削弱了YTHDF2與CNOT1(介導RNA降解的關鍵因子)的互作,并增強了與磷酸酶亞基MYPT1的互作,從而解釋了其功能失活。同時,UVB通過激活自噬降解YTHDF2蛋白,這一過程依賴于自噬受體p62。敲低自噬關鍵基因ATG5/7或p62,均可阻止UVB引起的YTHDF2下調及U6累積。這些發(fā)現(xiàn)闡明了環(huán)境應激如何通過破壞YTHDF2的穩(wěn)定性和功能,導致U6累積并最終引發(fā)炎癥。

圖6 UVB irradiation抑制YTHDF2活化
7、YTHDF2通過抑制TLR3通路抑制皮膚腫瘤發(fā)生
研究最終將分子機制與腫瘤發(fā)生相聯(lián)系。功能實驗表明,YTHDF2敲低促進皮膚癌細胞增殖、遷移和裸鼠移植瘤生長,而過表達則抑制這些表型。在慢性UVB誘導的小鼠皮膚癌模型中,皮膚特異性敲除YTHDF2顯著增加了腫瘤數(shù)量并加速了腫瘤發(fā)生。機制上,U6敲低或TLR3抑制均可逆轉YTHDF2敲低帶來的促瘤效應。同樣,使用COX-2抑制劑Celecoxib也能抑制YTHDF2敲低細胞的增殖。對人類皮膚鱗癌組織的分析顯示,YTHDF2蛋白水平顯著降低,且與腫瘤進展負相關。這些結果確立了YTHDF2通過抑制U6-TLR3軸在皮膚腫瘤發(fā)生中扮演關鍵抑癌角色。

圖7 YTHDF2通過抑制TLR3通路抑制皮膚腫瘤發(fā)生
全文結論:
研究揭示了一條由METTL16–m?A U6–YTHDF2–TLR3構成的全新信號軸,闡明了YTHDF2通過識別m?A修飾的內源性U6 snRNA并促進其降解,進而競爭性抑制U6與TLR3結合,從而在生理狀態(tài)下抑制炎癥反應的核心機制。研究進一步發(fā)現(xiàn),UVB輻射通過誘導YTHDF2去磷酸化及p62介導的自噬降解雙重途徑破壞該調控軸,導致U6累積并激活TLR3信號,最終驅動皮膚炎癥與腫瘤發(fā)生。體內外實驗證實YTHDF2具有抑制腫瘤的生物學功能,其表達在人類皮膚癌及自身免疫病中顯著下調。該發(fā)現(xiàn)不僅拓展了m?A修飾與非編碼RNA在免疫調控中的功能認知,也為相關疾病的治療提供了潛在新靶點。
研究意義與展望
本研究具有重要的理論與臨床意義。在理論層面,它突破了以往對m?A功能集中于mRNA的認知,揭示了一種由m?A修飾的“自我"非編碼RNA(U6 snRNA)驅動的先天性免疫激活新范式,將表觀轉錄組學、非編碼RNA生物學與免疫學緊密連接。其闡明的 YTHDF2作為“分子開關",通過競爭性結合抑制TLR3過度激活的機制,為理解細胞內如何區(qū)分并控制“自我"RNA的免疫原性提供了全新框架。
在臨床層面,該研究為炎癥性皮膚病、自身免疫疾病(如SLE)及皮膚癌的防治提供了新的潛在靶點。YTHDF2、METTL16或m?A U6本身,以及其下游的TLR3信號通路,均可作為潛在的干預窗口。例如,開發(fā)小分子激動劑以增強YTHDF2功能,或使用中和抗體阻斷U6與TLR3的結合,可能為緩解病理性炎癥和抑制腫瘤發(fā)生提供新策略。
此外,幾個關鍵問題有待深入探索:首先,m?A U6在其它組織、炎癥類型及癌癥中的作用是否普適?其次,YTHDF2的磷酸化調控網(wǎng)絡及其與自噬降解的串擾機制仍需細化。此外,能否開發(fā)出靶向該通路的安全有效療法,并克服TLR通路調控可能帶來的脫靶效應,是邁向臨床轉化的核心挑戰(zhàn)。總之,這項工作開辟了一個充滿前景的研究方向,預示著靶向RNA修飾及其閱讀蛋白可能成為未來免疫代謝疾病治療的新前沿。

![]()
技術支持:環(huán)保在線 地址:上海市寶山區(qū)長江南路180號B區(qū)B650 管理登陸 sitemap.xml
 微信二維碼
微信二維碼消息")
