-
巨噬細胞分泌CCL24通過成纖維細胞CCR3 促進心臟纖維化
發(fā)布時間: 2025-10-17 點擊次數(shù): 12次2025年8月21日,《Circulation Research》在線發(fā)表明尼蘇達大學Xavier S. Revelo團隊的突破性成果。研究揭示CCL24通過其受體CCR3和PI3K/ AKT信號的下游激活直接激活成纖維細胞,纖維細胞中CCR3的遺傳消融可改善心臟功能,并在壓力過載后改善纖維化。這項研究表明,CCL24/CCR3途徑的激活在促進纖維化和心臟功能障礙的響應中對壓力超負荷有關鍵作用。

背景:
炎癥是心血管疾病的重要危險因素,通過驅(qū)動對心臟損傷的適應性和非適應性反應,促進心血管疾病的發(fā)展。巨噬細胞是心臟中zui豐富的免疫細胞,在心臟組織重塑中發(fā)揮重要作用。心臟駐留巨噬細胞是心肌的重要組成部分,在炎癥反應、組織損傷和重塑中發(fā)揮關鍵作用。然而,心臟駐留巨噬細胞調(diào)節(jié)心力衰竭重塑的確切機制仍然知之甚少。
本文即在這一背景下,研究了CCL24在急性壓力超負荷引起的心力衰竭進展中的作用。遺傳缺陷和抗體介導的CCL24抑制導致心臟功能改善,適應性心臟重塑以及壓力過載后的纖維化改善。

關鍵詞
纖維化;心力衰竭;巨噬細胞;CCL24;M2巨噬細胞。
研究結果
結論1: CRMs是心臟中Ccl24的主要來源
研究發(fā)現(xiàn),心臟駐留巨噬細胞(CRMs)是CCL24的主要來源。通過單細胞測序、qPCR、熒光原位雜交和骨髓移植實驗證實,CCL24表達主要集中于CRMs,而非單核細胞來源巨噬細胞或其他心臟細胞。在壓力超負荷(TAC)模型中,CCL24水平在心臟重塑早期短暫升高,與CRMs擴增同步。IL-4與IL-10協(xié)同誘導CRMs產(chǎn)生CCL24,提示其在早期心臟重塑中通過CCL24介導促纖維化作用。
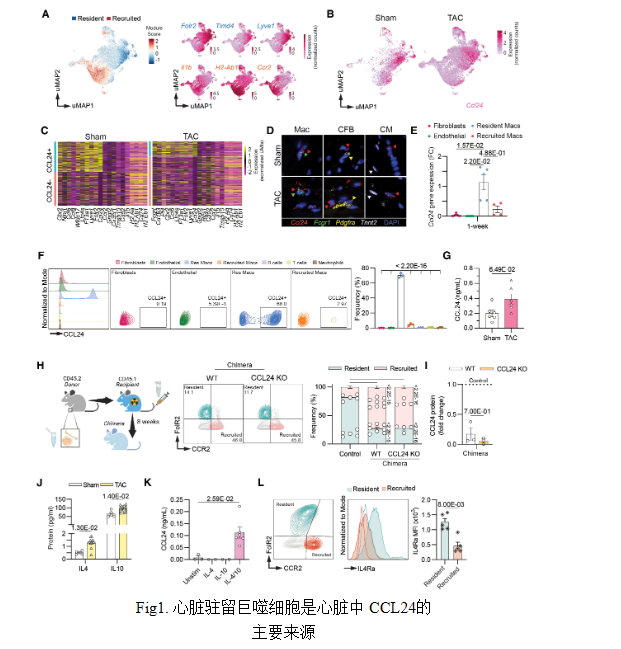
結論2: CRM來源的Ccl24促進收縮功能障礙。
壓力超負荷下,CCL24缺失或抗體中和均顯著改善小鼠收縮功能,抑制心室擴張,且肥大反應短暫。CCL24由CRM分泌,非MoMF來源,其存在阻礙心臟適應性重構,提示阻斷CCL24可成為心衰早期干預新策略。

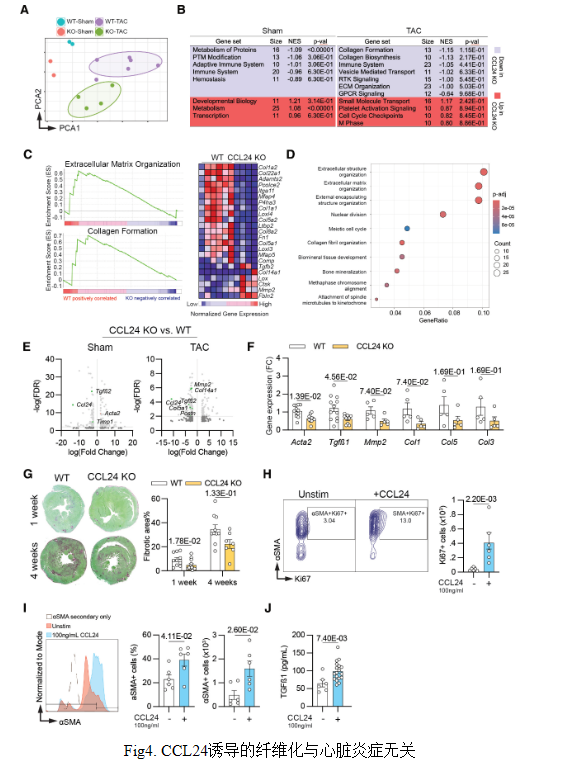
結論3:CCL24缺失可防止心臟纖維化,而CCL24在體外可激活成纖維細胞。
RNA-seq顯示,CCL24缺失下調(diào)膠原、ECM形成及TGFβ通路,促纖維化基因表達減少,心臟纖維化面積降40%。重組CCL24可直接刺激成纖維細胞增殖、αSMA及TGFβ產(chǎn)生,證實CCL24通過激活成纖維細胞驅(qū)動心臟纖維化。
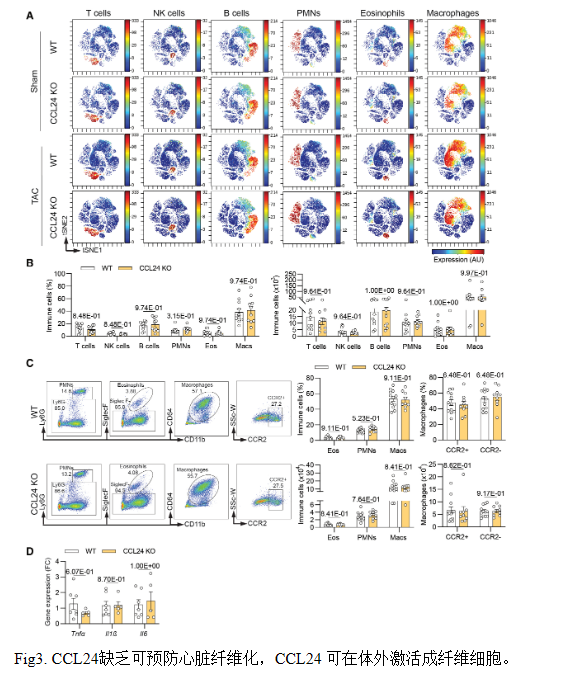
結論4: CCL24 誘導的纖維化重塑與心臟炎癥無關
CCL24缺失不改變穩(wěn)態(tài)或壓力負荷后心臟各免疫細胞亞群數(shù)量及炎癥基因表達,提示其促纖維化與心功能障礙作用獨立于炎癥細胞浸潤變化。
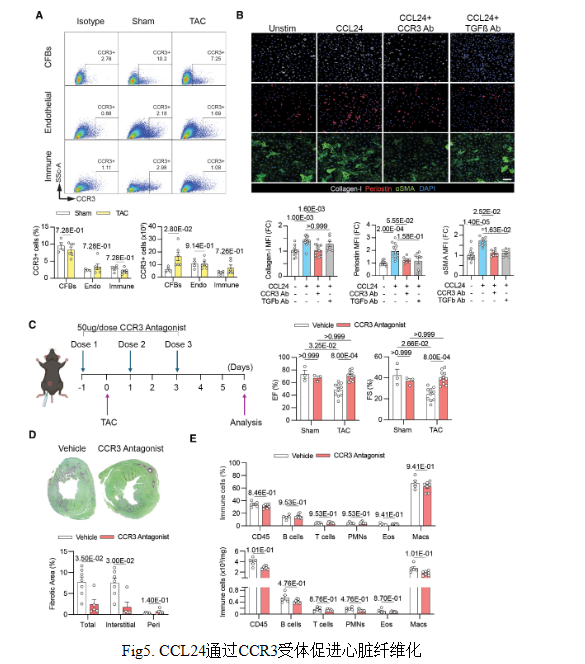
結論5: CCL24 通過 CCR3 受體促進纖維化
CCL24通過受體CCR3直接激活心臟成纖維細胞,誘導TGF-β依賴的膠原、αSMA表達,驅(qū)動纖維化;阻斷CCR3或TGF-β均可抑制該過程,改善心臟功能與間質(zhì)纖維化,證實CCL24-CCR3軸為可靶向的促纖維化通路。
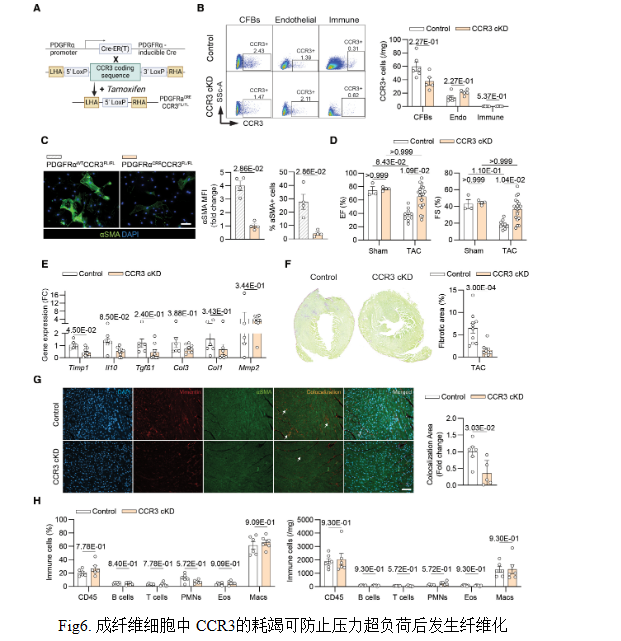
結論6:成纖維細胞中CCR3的cKD可防止纖維化發(fā)展
構建成纖維細胞條件性CCR3敲除小鼠(CCR3 cKD),他mo昔芬誘導后CCR3表達顯著下降。TAC術后1周,CCR3 cKD小鼠射血分數(shù)和縮短分數(shù)提高,Col1、Col3、Timp1等纖維化基因下調(diào),膠原沉積與肌成纖維細胞面積減少,心功能改善且不依賴免疫浸潤變化,證實成纖維細胞CCR3介導CCL24促纖維化作用。
結論7:CCL24 介導的 CCR3 激活通過 PI3K/Akt 途徑驅(qū)動心臟纖維化
CCL24經(jīng)CCR3觸發(fā)PI3K-Akt信號:刺激30–60分鐘磷酸化Akt,PI3K抑制劑或CCR3缺失均阻斷αSMA上調(diào);直接激活PI3K可繞過CCL24誘導肌成纖維細胞活化,證實PI3K-Akt為CCL24-CCR3促纖維化核心通路。
 。
。總結
本研究發(fā)現(xiàn),心臟駐留巨噬細胞在壓力超負荷下通過分泌CCL24激活成纖維細胞CCR3受體,經(jīng)PI3K/Akt通路促進TGF-β釋放與纖維化,加重心功能障礙;基因或藥物阻斷CCL24-CCR3軸顯著抑制纖維化并改善心功能,提出靶向該軸作為心衰早期抗纖維化新策略,但尚需驗證長期干預效果與安全性。
參考文獻:
Parthiban P, Barrow F, Wang H, et al. Macrophage-Derived CCL24 Promotes Cardiac Fibrosis Via Fibroblast CCR3. Circ Res. Published online September 16, 2025. doi:10.1161/CIRCRESAHA.125.326599

![]()
技術支持:環(huán)保在線 地址:上海市寶山區(qū)長江南路180號B區(qū)B650 管理登陸 sitemap.xml
 微信二維碼
微信二維碼消息")
